Home
NEWS
Quantum information theory on sparse wavefunctions and applications for Quantum Chemistry
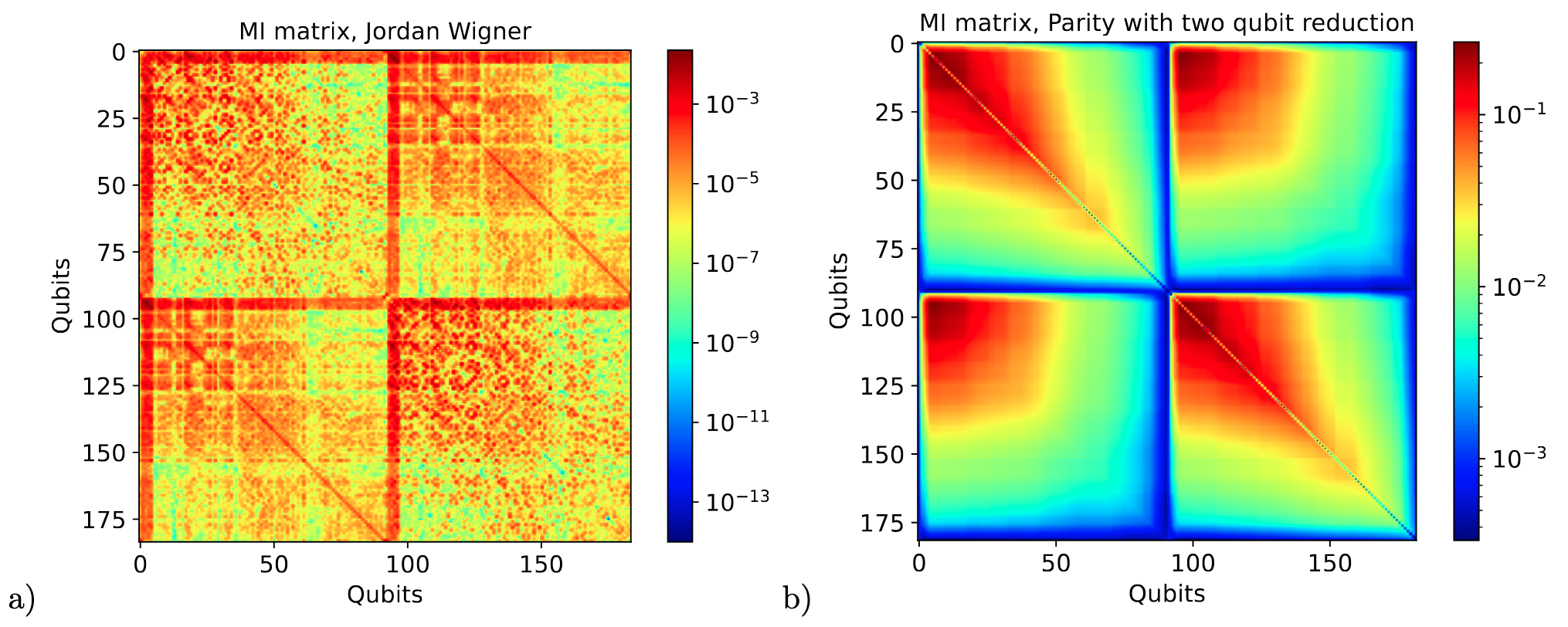
(Fig.1 Mutual information for the water molecule at CISD level. Jordan Wigner mapping is reported on the left panel, parity mapping with the two-qubit reduction on the right panel. The starting wavefunction for the mutual information was the simulation for the water molecule(H2O) )
ABSTRACT:
In recent years Quantum Computing prominently entered in the field of Computational Chemistry, importing and transforming computational methods and ideas originally developed within other disciplines, such as Physics, Mathematics and Computer Science into algorithms able to estimate quantum properties of atoms and molecules on present and future quantum devices. An important role in this contamination process is attributed to Quantum Information techniques, having the twofold role of contributing to the analysis of electron correlation and entanglements and guiding the construction of wavefunction variational ansatzes for the Variational Quantum Eigensolver technique.
This paper introduces the tool SparQ (Sparse Quantum state analysis), designed to efficiently compute fundamental quantum information theory observables on post-Hartree-Fock wavefunctions sparse in their definition space. The core methodology involves mapping fermionic wavefunctions to qubit space using fermionic-to-qubits transformations and leveraging the sparse nature of these wavefunctions to evaluate observables and properties of the wavefunction.
The effectiveness of SparQ is validated by analyzing the mutual information matrices of wavefunctions for the water molecule and the total entropy of qubits describing the benzene molecule. This highlights its capability to handle large-scale quantum systems, limited mainly by the capabilities of quantum chemical methods used to retrieve the wavefunctions. The results indicate that quantum information theoretical analysis, so far limited to traditional tensor network methods and study of transition operators, can be applied to all post-Hartree-Fock wavefunctions, extending their applications to larger and more complex chemical systems.
Authors: Davide Materia, Leonardo Ratini, Leonardo Guidoni
Publication date: 9 Sep 2025
Journal: Journal of Physical Chemistry A
DOI (full article): https://pubs.acs.org/doi/10.1021/acs.jpca.5c02137
- Written by Super User
- Hits: 217
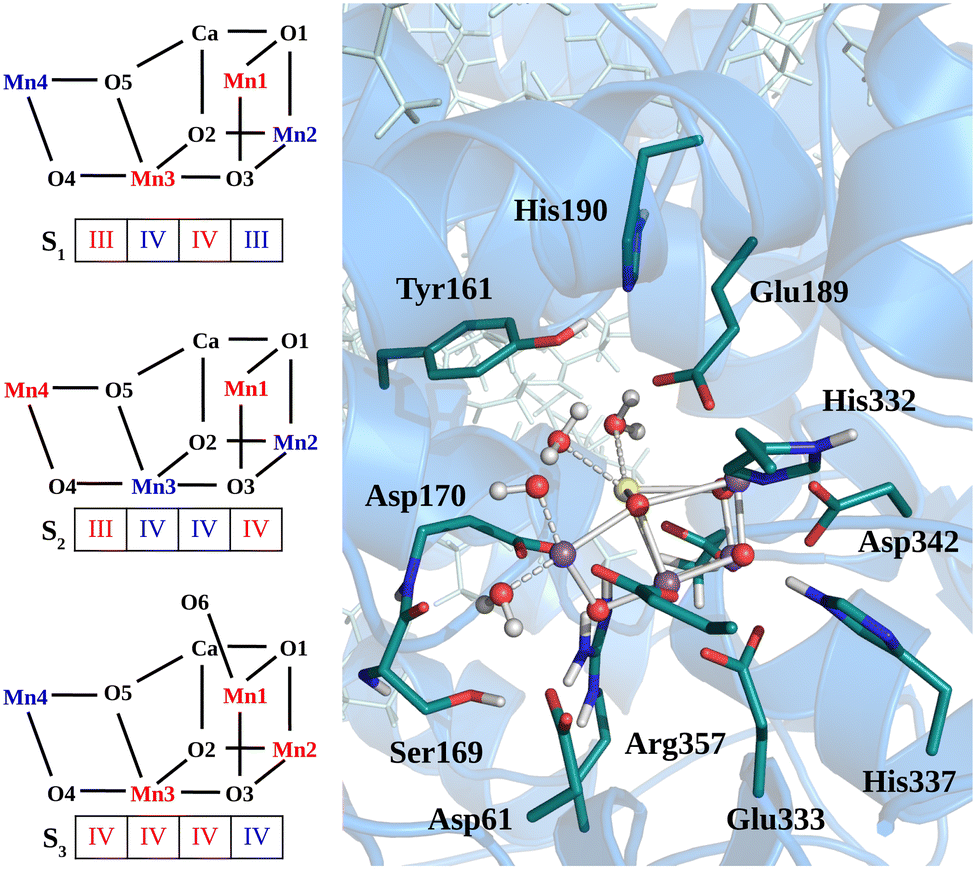
(Fig.1 Structural sketch of Mn4Ca cluster and the respective spin/redox for each studied state are shown in the left panel. In the right panels are shown the residues included in the QM region for all the QM/MM simulations. )
ABSTRACT:
Vibrational spectroscopy serves as a powerful tool for characterizing intermediate states within the Kok–Joliot cycle. In this study, we employ a QM/MM molecular dynamics framework to calculate the room temperature infrared absorption spectra of the S1, S2, and S3 states via the Fourier transform of the dipole time auto-correlation function. To better analyze the computational data and assign spectral peaks, we introduce an approach based on dipole–dipole correlation function of cluster moieties of the reaction center. Our analysis reveals variation in the infrared signature of the Mn4Ca cluster along the Kok–Joliot cycle, attributed to its increasing symmetry and rigidity resulting from the rising oxidation state of the Mn ions. Furthermore, we successfully assign the debated contributions in the frequency range around 600 cm−1. This computational methodology provides valuable insights for deciphering experimental infrared spectra and understanding the water oxidation process in both biological and artificial systems.
Authors: Capone Matteo, Parisse Gianluca, Narzi Daniele, Guidoni Leonardo
Publication date: 19 Jul 2024
Journal: Physical Chemistry Chemical Physics 2024
DOI (full article): https://doi.org/10.1039/D4CP01307G
- Written by Super User
- Hits: 6034
Natural orbitals and sparsity of quantum mutual information

(Fig.2 Representation of the H2O molecular orbitals in the frozen core approximation optimized by the algorithm. Each column corresponds to a different basis set: the first one represents the Hartree-Fock orbitals, the second one the WAHTOR-optimized orbitals and the one the natural orbitals.)
ABSTRACT:
Natural orbitals, defined in electronic structure and quantum chemistry as the (molecular) orbitals diagonalizing the one-particle reduced density matrix of the ground state, have been conjectured for decades to be the perfect reference orbitals to describe electron correlation. In the present work we applied the Wavefunction-Adapted Hamiltonian Through Orbital Rotation (WAHTOR) method to study correlated empirical ansätze for quantum computing. In all representative molecules considered, we show that the converged orbitals are coinciding with natural orbitals. Interestingly, the resulting quantum mutual information matrix built on such orbitals is also maximally sparse, providing a clear picture that such orbital choice is indeed able to provide the optimal basis to describe electron correlation. The correlation is therefore encoded in a smaller number of qubit pairs contributing to the quantum mutual information matrix.
Authors: Leonardo Ratini, Chiara Capecci, Guidoni Leonardo
Publication date: n/a (Accepted)
Journal: Journal of Chemical Theory and Computation
DOI (full article): https://doi.org/10.48550/arXiv.2308.08056
- Written by Super User
- Hits: 8076
The electron–proton bottleneck of photosynthetic oxygen evolution

(picture by Ph.D Matteo Capone)
ABSTRACT:
Photosynthesis fuels life on Earth by storing solar energy in chemical form. Today’s oxygen-rich atmosphere has resulted from the splitting of water at the protein-bound manganese cluster of photosystem II during photosynthesis. Formation of molecular oxygen starts from a state with four accumulated electron holes, the S4 state—which was postulated half a century ago1 and remains largely uncharacterized. Here we resolve this key stage of photosynthetic O2 formation and its crucial mechanistic role. We tracked 230,000 excitation cycles of dark-adapted photosystems with microsecond infrared spectroscopy. Combining these results with computational chemistry reveals that a crucial proton vacancy is initally created through gated sidechain deprotonation. Subsequently, a reactive oxygen radical is formed in a single-electron, multi-proton transfer event. This is the slowest step in photosynthetic O2 formation, with a moderate energetic barrier and marked entropic slowdown. We identify the S4 state as the oxygen-radical state; its formation is followed by fast O–O bonding and O2 release. In conjunction with previous breakthroughs in experimental and computational investigations, a compelling atomistic picture of photosynthetic O2 formation emerges. Our results provide insights into a biological process that is likely to have occurred unchanged for the past three billion years, which we expect to support the knowledge-based design of artificial water-splitting systems.
Authors: Greife Paul, Schönborn Matthias, Capone Matteo, Assunção Ricardo, Narzi Daniele, Guidoni Leonardo, Dau Holger
Publication date: 2023/05/03
Journal: Nature
DOI (full article): https://doi.org/10.1038/s41586-023-06008-5

Cited on:
Nature's News and View
ANSA.it article
Univaq Official Press release (pdf)
- Written by Super User
- Hits: 11142

(Fig.2 WAHTOR Algorithm scheme)
ABSTRACT:
By exploiting the invariance of the molecular Hamiltonian by a unitary transformation of the orbitals it is possible to significantly shorter the depth of the variational circuit in the Variational Quantum Eigensolver (VQE) algorithm by using the Wavefunction Adapted Hamiltonian Through Orbital Rotation (WAHTOR) algorithm. This work introduces a non-adiabatic version of the WAHTOR algorithm and compares its efficiency with three implementations by estimating Quantum Processing Unit (QPU) resources in prototypical benchmarking systems. Calculating first and second-order derivatives of the Hamiltonian at fixed VQE parameters does not introduce a significant QPU overload, leading to results on small molecules that indicate the non-adiabatic Newton-Raphson method as the more convenient choice. On the contrary, we find out that in the case of Hubbard model systems the trust region non-adiabatic optimization is more efficient. The preset work therefore clearly indicates the best optimization strategies for empirical variational ansatzes, facilitating the optimization of larger variational wavefunctions for quantum computing.
Authors: Leonardo Ratini, Chiara Capecci, Guidoni Leonardo
Publication date: 27 October 2023
Journal: Electronic Structure
DOI (full article): https://dx.doi.org/10.1088/2516-1075/ad018e
- Written by Super User
- Hits: 9703